

Local sequencing efforts reveal patterns in COVID-19 variant evolution
A team of researchers has recently shown seven independent virus variant lineages across the United States that have evolved a mutation in the same place on the SARS-CoV-2 spike protein. Local sequencing company Microbial Genome Sequencing Center did the sequencing for this study.
April 21, 2021
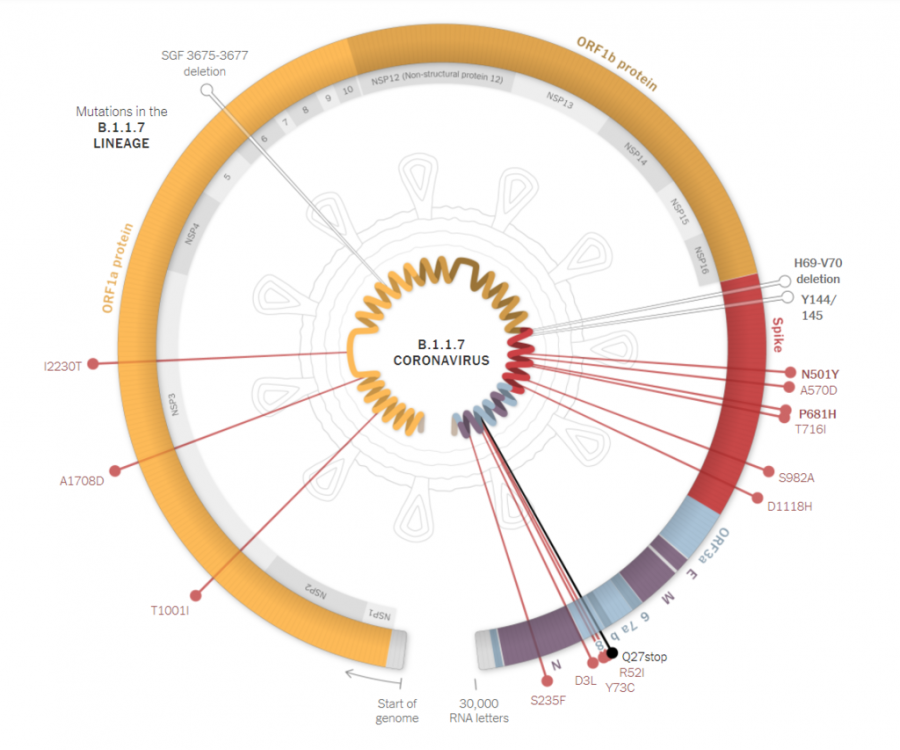
As COVID-19 continues to spread, new variants of the virus are being discovered and researched. One of these variants — B.1.1.7, which was first identified in the United Kingdom — was detected on Pitt’s campus last month.
A team of researchers has recently shown seven independent virus variant lineages across the United States, which have evolved a mutation in the same place on the SARS-CoV-2 spike protein. The sequencing for this study was done through local sequencing company Microbial Genome Sequencing Center, which was co-founded by Vaughn Cooper, a microbiology professor at Pitt and director of the Pittsburgh Center for Evolutionary Biology and Medicine.
The recent preprint on medRxiv, an online server of health science research that has not yet been peer-reviewed for journal publication, came out on Feb. 14. It describes convergent evolution of variants found in the United States. Jeremy Kamil, virologist at Louisiana State University Health Sciences Center Shreveport and a co-author of the study, was instrumental in collecting the samples that MiGS sequenced for this study.
“A variant is like a unique constellation of mutations while a mutation will be like one star in that constellation,” Kamil said. “Alternatively, if a variant was a fingerprint, then a mutation will be one line that’s part of the fingerprint.”
Kamil said the biggest reason why there are so many mutations in the SARS-CoV-2 virus, which causes COVID-19, is because the pandemic has not been controlled and when there are high numbers of virus replication events, there are more chances for errors.
“The more out of control the pandemic, the more chances for some random mutation to be favored by selection,” Kamil said.
The COVID-19 pandemic has offered a unique chance to study viral evolution. Kamil said the evolutionary conclusions could be made because having so many samples helps researchers get closer to finding out what mutations are truly meaningful.
While this study focused on the convergent evolution of mutations in position 677 of the COVID-19 spike protein, Kamil said there are variants whose mutation cluster is in the range of 675 to 681, which supports the important functionality of this section of the spike protein. Kamil said he suspects it is from adaptation to the human respiratory tract. Kamil said follow-up studies are already being conducted to better understand how mutations in this region contribute to SARS-CoV-2’s fitness.
According to Cooper, what is remarkable about this study is not just the evolutionary finding, but also the accelerated timeline from data to paper. Compared to the usual timeline for research from data collection to paper, the combination of high volume of SARS-CoV-2 samples, MiGS’ sequencing power and preprint popularity has greatly accelerated the publication timeline from an average two years to two weeks.
“We did it so fast, Jeremy shared the timeline of our discovery and it was just unbelievable. It was from Jan. 27 to Feb. 14, from the first inkling to a medRxiv solid paper,” Cooper said.
Dan Snyder, the director of MiGS and author on the study, said understanding SARS-CoV-2’s evolution and functions of its spike protein is still critical going forward, especially as people around the world get vaccinated.
“I’m curious to see what will happen as the selective pressure of the vaccine starts to bear down on the circulating virus population,” Snyder said.
Snyder said there is already an ongoing project at MiGS in collaboration with Dr. Lee Harrison, head of the School of Medicine’s Infectious Diseases Epidemiology Research Unit, to sequence bacterial infection samples and investigate trends of UPMC patient infections.
Cooper said sequencing centers like MiGS are well poised to expand the application of genome sequencing and have already showcased how microbial genome sequencing and surveillance can be used as a public health tool.
“I’ve always thought that genome-based surveillance was important in health care because it can detect transmission events between patients. Sequencing thousands of potential bacterial genomes and trying to identify chains of transmission, it actually saves money,” Cooper said.
Kamil said genome-based surveillance could become a public health tool for viruses, bacteria and fungi in the community. He said local sequencing efforts combined with regular sampling in both hospitals and public spaces could be used as a weather report for pathogens circulating in the community. This could better inform doctors in their treatment plans and protect our communities from future pandemics.
“The cost to have constant sequencing is a drop in the bucket compared to the damage that a pandemic costs,” Kamil said.
News coverage of variants has been common throughout this pandemic. Cooper said people get nervous that there are all these different variants out there, but just because there’s a different collection of mutations doesn’t mean that it functionally changed anything about the virus. Understanding the relative importance of certain mutations and continued research on SARS-CoV-2’s evolution is “important for calming mania about variants,” according to Cooper. But Cooper said most importantly, we need to stop infections with continued social distancing and safety protocols.
“More infections equals more opportunity for the virus to adapt. So if you are trying to preserve the global public health good, you have to stop the fire,” Cooper said.
